Atlas of Cardiac Genetic Variation
The Atlas of Cardiac Genetic Variation was produced from a collaboration between the labs of Professor Stuart Cook (National Heart and Lung Institute, Imperial College London and NIHR Cardiovascular Biomedical Research Unit, Royal Brompton Hospital) and Professor Hugh Watkins (Radcliffe Department of Medicine, University of Oxford and Oxford Molecular Genetics Laboratory, Oxford University Hospitals), as part of our publication "Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples" (Genet Med. 2016 doi:10.1038/gim.2016.90)..
Background
The genetics of cardiomyopathy has become increasingly heterogeneous and complex in recent years, with ever more genes and variants associated with each condition. However, as our understanding of the level of rare variation in the general population has grown, it has become clear that many of these associations are not robust and may in fact represent benign, background variation unconnected to disease. The Atlas of Cardiac Genetic Variation utilises two substantial resources of genetic data - population data from the Exome Aggregation Consortium (ExAC) and clinical data from the Oxford Molecular Genetics Laboratory (OMGL) and the Laboratory of Molecular Medicine (LMM) - to clarify the genetics of cardiomyopathies and inform clinical decision making.
Exome Aggregation Consortium
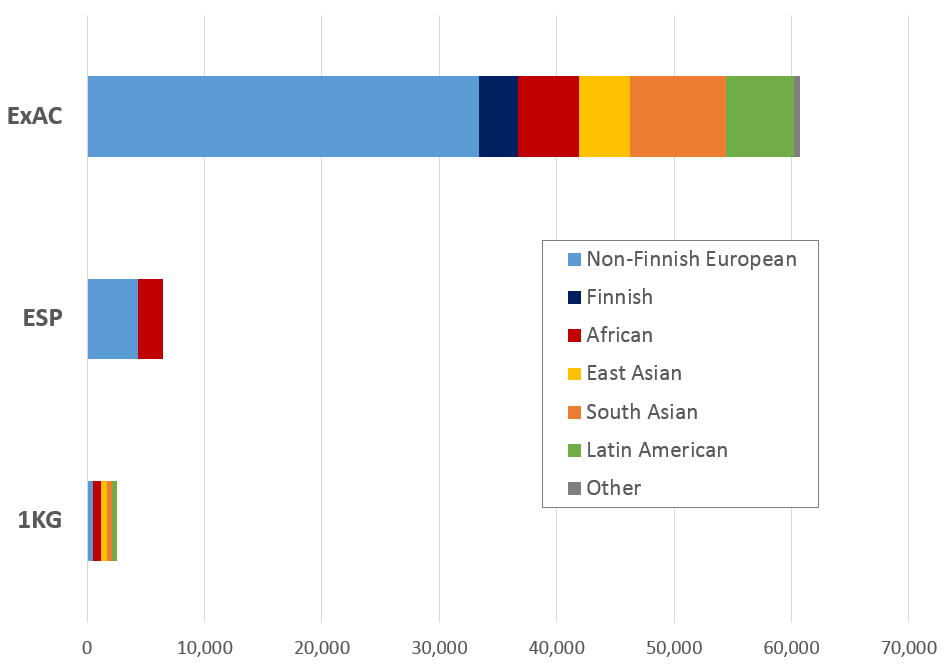
The Exome Aggregation Consortium (ExAC) is a coalition of investigators seeking to aggregate and harmonise exome sequencing data from a variety of large-scale sequencing projects, and to make summary data available for the wider scientific community. Developed at the Broad Institute and first released in October 2014, ExAC currently comprises 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies.
The scale of ExAC is an order of magnitude greater than previous population databases (Exome Sequencing Project - ESP, 1000 Genomes - 1KG, right) and is the first dataset powered to assess variant alleles present in the population at a range of 1:1000-100000 that might have previously been considered pathogenic but may in fact be too common to cause penetrant Mendelian disease. ExAC also enables the accurate calculation of the level of rare variation across genes, in particular for highly constrained genes, allowing for case-control studies to assess the validity of gene associations with Mendelian disease.
For more details on the generation and composition of ExAC, see the ExAC FAQ.
Clinical cardiomyopathy cases
The 7,855 clinical cardiomyopathy cases assessed in this study are the product of over 10 years of clinical genetic testing in the OMGL (Oxford, UK) and LMM (Boston, USA) laboratories. The LMM data was published in two paper in 2014-15 (references below) while the OMGL was published for the first time in this study. The cases comprised:
- Hypertrophic Cardiomyopathy (HCM) - 6179 cases (3267 from OMGL, 2912 from LMM)
- Dilated Cardiomyopathy (DCM) - 1315 cases (559 from OMGL, 756 from LMM)
- Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) - 316 cases (all from OMGL)
References for LMM data:
Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH.
The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing.
Genet Med. 2014 Aug;16(8):601-8.
Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, Cox SW, DePalma SR, Ho CY, Seidman JG, Seidman CE, Rehm HL. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015 Nov;17(11):880-8.
Case excess
A case excess is the amount of rare variation in a gene observed in a cohort of patients with a particular disease over and above a control or reference population cohort. It is defined as: proportion of cases with a rare variant in a gene - proportion of controls/reference samples with a rare variant. For most genes with a valid association to Mendelian diseases like cardiomyopathies, a statistically significant excess of rare variants will be observed. While a lack of case excess does not necessarily preclude a gene from a pathogenic role in disease (genes may be very rare causes of disease with pathogenic variants limited to small regions of the gene), variants in these genes are likely to be uninterpretable without substantial evidence of segregation in the affected family and therefore of limited utility for clinical genetic testing.
Variant interpretation - Odds Ratios and Etiological Fractions
A comprehensive assessment of the level of rare variation across genes in the population (ExAC) and large case cohorts allows us to calculate metrics
to evaluate the pathogencity of novel variants when detected in a proband. These are the Odds Ratio (OR) (ratio of odds of cardiomyopathy comparing rare variant
carriers with non-carriers) and the Etiological Fraction (EF) (an estimation of the proportion of cases in which the rare variant in
a gene is causal). Values are calculated for truncating (frameshift, nonsense, canonical splice site) and non-truncating (missense, inframe indel)
variants for each gene.
e.g. a rare truncating variant in MYBPC3 in a HCM proband has an EF of 0.99 and an OR of 119,
whereas a rare non-truncating variant in MYBPC3 in a HCM proband has an EF of 0.82 and an OR of 5.7.
For more details about this study and how the data was generated, please see our paper "Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples" (Genet Med. 2016 doi:10.1038/gim.2016.90)..