Paralogue Annotation for Inherited Arrhythmias
Paralogue annotation is a novel technique which aims to identify novel missense variants that are likely to disease-causing. The technique utilises known clinical genetic mutations in related genes/proteins (paralogues) and tranfers them to equivalent amino acids in your protein of interest, identifying residues likely to be intolerant to variation which may harbour novel missense variants. Detailed information on the paralogue annotation of disease-causing variants in Long QT syndrome, Brugada syndrome, CPVT and RYR1 myopathies is available here.
See below for tutorials on how paralogue annotation identifies novel pathogenic variants and how this website can be used by researchers and clinicians of inherited arrhythmias. For more information, read the following publications:
- Paralogous annotation of disease-causing variants in Long QT syndrome genes - Hum Mutat. 2012 Aug;33(8):1188-91 (concept and validation)
- Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia - J Med Genet doi:10.1136/jmedgenet-2013-101917 (application to clinical studies)
| LQT | Gene | LRG | Paralogue Mappings - Residues (Total Variants) |
|---|---|---|---|
| Long QT syndrome | |||
| LQT1 | KCNQ1 | LRG_287 | 157 (248) |
| LQT2 | KCNH2 | LRG_288 | 130 (173) |
| LQT3 | SCN5A | LRG_289 | 712 (1194) |
| LQT4 | ANK2 | LRG_327 | 15 (15) |
| LQT5 | KCNE1 | LRG_290 | 17 (21) |
| LQT6 | KCNE2 | LRG_291 | 39 (43) |
| LQT7 | KCNJ2 | LRG_328 | 144 (234) |
| LQT8 | CACNA1C | LRG_334 | 852 (1609) |
| Brugada syndrome | |||
| BrS1 | SCN5A | LRG_289 | 712 (1194) |
| Ryanodine receptors | |||
| CPVT1 | RYR2 | LRG_402 | 380 (444) |
| RYR1 | 229 (260) | ||
How Paralogue Annotation Works

For your gene of interest (in this example SCN5A), identify all similar human genes (paralogues) by sequence homology and check whether a subset of these genes are involved in Mendelian disease with known missense variants (using HGMD database). Here, two of these paralogues are displayed - SCN1A (linked to epilepsy and Dravet syndrome) and CACNA1A (linked to migraine and episodic ataxia).
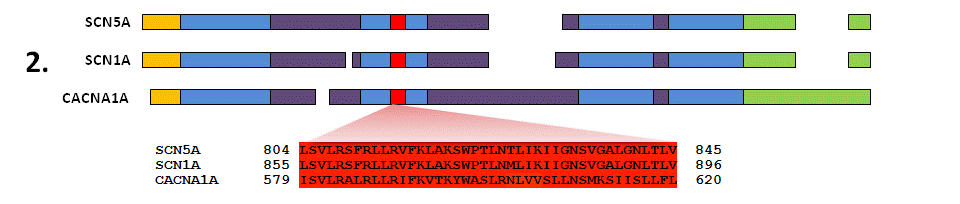
Align the protein sequences of each member of this gene family to map related regions and amino acids together. This will correlate the equivalent amino acids between all the proteins in the analysis and assess how similar the protein sequence and structure is at every amino acid residue.
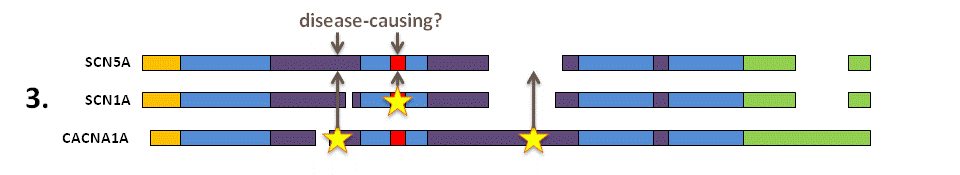
Identify the disease-causing missense variants in the aligned paralogues and the equivalent amino acids in your protein of interest. Novel missense variants altering these amino acids in SCN5A are predicted to have a similar effect on molecular function as in the paralogue proteins and therefore are likely to be the disease-causing mutation in this patient.
Using this website to classify novel variants with paralogue annotation
This website can be used to analyse novel variants for paralogue annotation across genes involved in Long QT syndrome (LQTS), the major genes of Brugada syndrome (SCN5A) and CPVT (RYR2), as well as RYR1-associated myopathies. After inputting the gene and amino acid/cDNA coordinates to the form above, you will be directed to the specific amino acid residue page. If paralogue annotation is available for this residue, the information can be interpreted as follows:

1. For this amino acid residue (RYR2 residue 357), mutations in the equivalent amino acid in RYR1 (G341R) have been linked to malignant hypothermia in several reports. The mapping quality of the equivalent amino acids is "High", indicating both proteins have the same reference amino acid and there is high homology between these proteins at this position.
2. A section of the multiple sequence alignment of RYR2 and its paralogues is shown, shaded according to T/M-coffee consensus scores (0-9). Users can check the strength of the alignment and homology between the paralogues, particularly at the amino acid of your protein of interest and the paralogue with known disease-causing mutations. The closer the homology between these proteins, the more likely that alterations to the equivalent amino acids will have similar molecular and physiological effects.
3. Before any clinical application it is crucial that users evaluate the evidence for pathogencity in the original report for the paralogue variant. PubMed links are provided to facilitate this. Variants in the literature may be reported as disease-associated simply on the basis of an observation in a disease cohort, or may have been thoroughly characterised using functional assays or family segregation studies. It is also important to check that the functional effect of the variant in the paralogue and gene of interest are equivalent - e.g. if your disease of interest is caused by gain of function, but the observed paralogue variant causes loss of function, then this annotation should be treated with caution.
References
-
Paralogous annotation of disease-causing variants in Long QT syndrome genes
Ware JS, Walsh R, Cunningham F, Birney E, Cook SA. Hum Mutat. 2012 Aug;33(8):1188-91 -
Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia
Walsh R, Peters NS, Cook SA, Ware JS. J Med Genet doi:10.1136/jmedgenet-2013-101917